

The inner ear vestibular system of marine reptiles and cetaceans adapted in similar ways to open-ocean swimming, a study finds.
Image credit: Bryan Christie Design.

Edited by Edward C Holmes, Pennsylvania State University, University Park, PA, and accepted by the Editorial Board October 14, 2012 (received for review February 27, 2012)
Determining the genetic pathways that viruses traverse to establish in new host species is crucial to predict the outcome of cross-species transmission but poorly understood for most host–virus systems. Using sequences encoding 78% of the rabies virus genome, we explored the extent, repeatability and dynamic outcome of evolution associated with multiple host shifts among New World bats. Episodic bursts of positive selection were detected in several viral proteins, including regions associated with host cell interaction and viral replication. Host shifts involved unique sets of substitutions, and few sites exhibited repeated evolution across adaptation to many bat species, suggesting diverse genetic determinants over host range. Combining these results with genetic reconstructions of the demographic histories of individual viral lineages revealed that although rabies viruses shared consistent three-stage processes of emergence in each new bat species, host shifts involving greater numbers of positively selected substitutions had longer delays between cross-species transmission and enzootic viral establishment. Our results point to multiple evolutionary routes to host establishment in a zoonotic RNA virus that may influence the speed of viral emergence.
Understanding how natural selection operates in novel or changing environments is important for managing a variety of ecological problems, including species responses to climate change and the dynamics of biological invasions. Evolutionary dynamics are particularly salient in pathogens such as RNA viruses, whose tendency to jump between host species makes them a major source of newly emerging infectious diseases affecting humans, domestic animals, and wildlife (1). The success of RNA viruses in crossing species barriers is enhanced by their high mutation rates (generated by error-prone RNA polymerases and large within-host population sizes), which provide genetic and phenotypic variability that enable ongoing transmission in new hosts (2). However, because RNA viruses have small genomes and multifunctional proteins, many potentially beneficial mutations have deleterious consequences for other aspects of viral fitness, making epistatic interactions a potentially important constraint on viral evolution (3, 4). This could reduce the number of evolutionary routes to host establishment or enforce fundamental limits on host range (5).
Evolutionary biologists have long portrayed adaptive change as a fitness landscape, with isolated peaks separated by valleys of lower fitness (6). For host-shifting viruses, an analogous landscape, where potential host species constitute distinct peaks, may emerge from the need for viruses to adapt to the physiological and ecological differences between donor and recipient host species (7). If adaptation requires few evolutionary changes, viral establishment should occur quickly, and disease control efforts might focus more on limiting initial cross-species transmission than on posttransmission measures. Alternatively, if establishing in novel hosts requires greater numbers of adaptive changes or suites of co-occurring or sequential changes, this could provide a longer window for intervention within populations of the newly infected host species. Empirical support exists for both pathways to viral establishment. For example, Venezuelan equine encephalitis required few adaptive changes to infect horses, whereas other host shifts [e.g., severe acute respiratory syndrome (SARS) in humans] were associated with extensive genetic change (8, 9). Predicting the evolutionary dynamics of future host shifts from single emergence events is challenging because the strength of barriers to establishment likely depend on both the host species and viral variant involved. Comparative data from multiple host shifts are therefore needed to assess whether molecular pathways to host establishment are repeatable and whether the extent of evolutionary change affects the speed of emergence.
Rabies virus (RV) (Lyssavirus, Rhabdoviridae), a zoonotic RNA virus with an evolutionary history dominated by host shifts within and among bats and carnivores, provides a rare opportunity to compare the evolutionary dynamics of repeated viral establishment (10). Host shifts are especially prominent among New World bats, many of which harbor species-specific viral lineages that share a relatively recent common ancestor (11). Previous studies investigated the determinants of Lyssavirus adaptation to new host species using the genomic distribution of amino acid sites under selection. These studies focused on the glycoprotein (G), the sole surface protein responsible for host cell interaction and entry into the nervous system, and the nucleoprotein (N), which forms the viral capsid and plays a role in transcription and replication. Highly localized amino acid changes were observed in the ectodomain of G, a region associated with host cell entry, but the mechanisms driving these changes were unclear (10, 12). Another possibility is that adaptation depends on changes in the RNA-dependent RNA polymerase [the large gene (L)]. This gene regulates viral transcription and replication, which could ultimately alter pathogenesis, virulence, and transmission, as observed in avian metapneumoviruses and paramyxoviruses (13, 14). However, no studies have investigated the role of L in the establishment of new RV reservoirs.
Here, we applied Bayesian phylogenetic ancestral state reconstruction to sequence data from the N, G, and L genes of 30 bat RV lineages to identify plausible host shifts between species. Combining these estimates of donor–recipient relationships with recently developed analyses of historical selection pressures allowed us to identify episodic positive selection in the RV genome and to compare the extent and repeatability of evolutionary changes associated with numerous host shifts. Finally, we used estimates of past viral demography derived from genetic data to investigate associations between the number of positively selected changes since viral introduction and the speed of establishment in each bat species.
We quantified the rates of nonsynonymous (dN) and synonymous (dS) substitutions across the branches of maximum likelihood (ML) phylogenetic trees inferred for the N, G, and L genes of major bat RV lineages from a total of 184 viral isolates. The relative difference in dN and dS indicates the selection pressure exerted on genes, with dN/dS > 1 (or equivalently, dN − dS > 0) indicative of positive selection. The overall dN/dS ratios were low (0.05, 0.15, and 0.05 in the N, G, and L genes, respectively), indicating a dominance of purifying selection when averaging across all sites and branches of the phylogenetic trees. Next, we examined selection on specific amino acid positions in two ways: first, we used a fixed effects likelihood (FEL) analysis that assumed constant selective pressure over time; and second, through a mixed effects model of episodic selection (MEME) to allow for temporally varying positive selection (15, 16). For all three genes, both methods identified amino acid sites putatively evolving under positive selection (Table S1). Nearly all sites detected by FEL were also supported by MEME; however, MEME further identified positively selected sites that were classified as evolving neutrally or under purifying selection by FEL (Table S1). These disagreements reflect the divergent assumptions of each model and are consistent with transient positive selection followed by enduring purifying selection to maintain those changes (15). Indeed, likelihood ratio tests (LRTs) comparing the MEME to the FEL models for individual sites commonly favored the episodic selection model over the constant selection model (Table S1). On average, positively selected codons in N, G, and L only experienced selection on 0.8%, 4.6%, and 2.6% of branches in their respective phylogenies. However, several codons underwent more frequent substitutions. For example, site G493 switched nine times between six different amino acid residues, G357 underwent seven flip-flop substitutions between valine and isoleucine, and L1620 switched repeatedly between glycine and four other residues (Fig. 1 B and C). In each of these sites, LRTs favored the FEL model over the more complex MEME, consistent with more pervasive selection over time (Table S1).
Episodic positive selection in the bat RV phylogeny. Maximum likelihood topologies of the bat RV N, G and L genes are shown in A, B and C, respectively. Symbols indicate amino acid changes in putatively positively selected sites only. Color spectra of points follow the relative position along each gene. Inset pie charts show the ratio of internal to tip substitutions for all sites in each gene. Lineage labels denote the reservoir host species as follows: Ap, Antrozous pallidus; Ct, Corynorhinus townsendii; Dr, Desmodus rotundus; Ef, Eptesicus fuscus; Efr, Eptesicus furinalis; Hm, Histiotus montanus; Ln, Lasionycteris noctivagans; Lb, Lasiurus borealis; Lc, Lasiurus cinereus; Le, Lasiurus ega; Li, Lasiurus intermedius; Ls, Lasiurus seminolus; Lx, Lasiurus xanthinus; Mm, Molossus molossus; M, Myotis sp.; Ma, Myotis austroriparius; Mc, Myotis californicus; Mn, Myotis nigricans; My, Myotis yumanensis; Nh, Nycticeius humeralis; Nl, Nyctinomops laticaudatus; Ph, Parastrellus hesperus; Ps, Perimyotis subflavus; Tb, Tadarida brasiliensis.
Positively selected sites were located mainly within the first 160 codons of N (Table S1). In contrast to G and L, the substitutions in N occurred almost exclusively on the tips of the tree, indicative of false positives with respect to host adaptation (Fig. 1 and Fig. S1). We therefore restricted further analyses of positive selection to G and L, where substitutions along internal branches were more consistent with a role in host establishment. In G, nine positively selected sites were found in the ectodomain, including a main antigenic site, and two others were found in the endodomain of G, which interacts with internal viral proteins (Table S1). We also found an elevated dN in site 333 of the ectodomain, a position of known importance for the attenuation of laboratory RV strains, although this did not meet our criteria of statistical significance (dN/dS = 1.189; PFEL = 0.81; PMEME = 0.07) (17). In L, positive selection was detected in all of the putative functional domains except region II (Table S1).
To characterize and quantify the positively selected changes associated with RV shifts into new bat species, we used ancestral state reconstruction to assign host states and genetic changes to the internal branches of a consensus phylogeny of RV estimated from a joint Bayesian analysis of the N, G, and L genes (SI Text and Fig. S2). Transfers into new host species were followed by substitutions at zero to five positively selected sites. Host shifts involved unique evolutionary routes, rather than frequent alteration of a few, key sites linked to host tropism (Fig. 1 and Fig. S2). Pairs of amino acid sites rarely underwent more substitutions on the same branch than expected by chance, and such epistatic interactions were particularly rare among positively selected sites (Table S2).
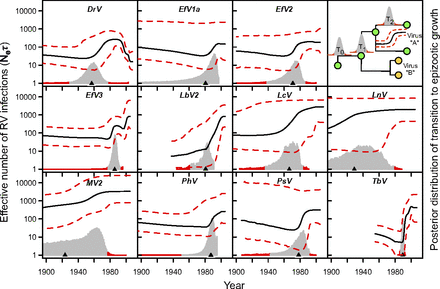
To characterize the epizootic dynamics of each host shift, we applied a Bayesian coalescent approach to infer past viral population dynamics. In 11 of the 13 RV lineages for which we had sufficient N gene sequence data for this analysis, we detected significant signatures of viral population growth (Table S3). In most of these lineages, reconstructions of past demographic histories using the nonparametric Bayesian skyline model revealed similar three-stage processes of host shifts. Nearly all RV lineages experienced a lag phase with a low effective number of infections, an epizootic phase during which infections increased, and an enzootic phase where the effective number of infections plateaued (Fig. 2).
Demographic histories and transition times to epizootic growth for 11 bat RV lineages. Each graph shows a Bayesian skyline plot estimated from N gene data. The effective number of infections is the product of the effective population size (Ne) and the generation time between infections (τ). Dashed lines are the 95% highest posterior density. Overlaid histograms show the posterior distributions of the transition times between historical (pregrowth) and contemporary demographic periods from the two-epoch models, with 95% limits shaded in red. Small black triangles are median transition times. The vertical axis for histograms was varied across graphs to enable comparable visualization, and the scale was therefore omitted. Top Right graph shows a schematic for calculating the epizootic lag time from the two-epoch models and the joint phylogeny. The epizootic lag time for virus A is T0 – T2, when including inferred ancestral-host states (scenario 2); or T1 – T2, when the origins of host shifts were treated as unknown (scenario 1).
We hypothesized that viruses might establish more quickly in new hosts when adaptation required changes in fewer numbers of positively selected sites, whereas longer lag phases might correspond to more extensive evolutionary change (7). To investigate the speed of viral emergence, we estimated the temporal delay between cross-species transmission and the onset of epizootic growth for each RV lineage. The date of cross-species transmission was estimated from the consensus bat RV phylogeny, assuming either that transmission occurred either: scenario 1, at the base of the “stem” branch of each RV lineage; or scenario 2, including additional branches of the RV phylogeny assigned to each host species by ancestral state reconstruction following a criterion of statistical significance (Fig. 2, Top Right graph and SI Text). The latter estimate is more biologically accurate because evolutionary changes on internal branches occurred in a bat species, but it suffers increased statistical uncertainty because ancestral host states could not always be assigned unambiguously, particularly in deeper nodes of the RV phylogeny (Table S4). The “epizootic lag time” was then calculated probabilistically as the difference between the posterior distribution of the year of cross-species transmission and the posterior distribution of the year of the transition between historical (small) and contemporary (large) effective numbers of viral infections according to Bayesian “two-epoch” demographic models (Fig. 2, Top Right graph). By iteratively regressing random draws from the posterior distribution of each epizootic lag time against the number of sites that underwent positively selected amino acid changes, we tested the relationship between the extent of molecular adaptation and delays in viral establishment, while accounting for phylogenetic and demographic uncertainty. Under scenario 2, greater numbers of amino acid changes in G and L were associated with longer delays until enzootic viral establishment, as evidenced by the lack of overlap of the 95% confidence interval (CI) of the slope parameter with zero (Fig. 3, slope; β: 95% CI = 10.71–79.96). The relationship was also significant when we considered only the number of changes in L (β: 95% CI = 14.02–116.54) but was less supported by data from G alone (β: 95% CI = −8.86–81.03). The more conservative dates of cross-species transmission and, therefore, shorter lag times (scenario 1 above), yielded similar results for L (β: 95% CI = 12.27–133.81), but a negative relationship emerged between lag time and the number of selected sites in G (β: 95% CI = −107.96 to −5.33), perhaps reflecting the greater frequency of changes in G on deeper branches of the phylogeny (Fig. 1) or a spurious effect arising from the low numbers of sites that underwent positively selected changes in this analysis.
Relationship between the number of positively selected amino acid changes in G and L and the epizootic lag time. Substitutions on terminal branches and in N were excluded because the timing of these substitutions was inconsistent with a postulated role in host adaptation. The black line and points show the median relationship with corresponding statistics in the Upper Left corner. Gray lines show 2,000 model predictions using random draws from the posterior distribution of epizootic lag time for each viral lineage. The inset histogram shows the distribution of the slope parameter from the iterated models, with 95% limits shaded in black (note the lack of overlap with zero).
Because the more abundant synonymous and nonpositively selected, nonsynonymous (NPN) substitutions largely determined the branch lengths that defined the epizootic lag time in this analysis, these substitutions were not surprisingly also correlated with the epizootic lag time in univariate tests (Fig. S3). However, neither was robust to inclusion in multivariate generalized linear models that contained positively selected sites, which remained highly significant (positively selected: F1,8 = 8.20, P = 0.021; synonymous: F1,8 = 1.47, P = 0.250; NPN: F1,8 = 3.11, P = 0.12). Moreover, models with positively selected sites alone provided a significantly better fit than models including either of the other substitution types according to Akaike's information criterion (versus synonymous: ΔAIC = 4.83; versus NPN: ΔAIC = 4.14). These differences in predictive power reflected the absence of strong correlations between the numbers of positively selected and synonymous substitutions (r = 0.56; P = 0.071) or NPN substitutions (r = 0.50; P = 0.12), suggesting that positively selected changes do not simply and necessarily accumulate as a function of time.
The repeated emergence of RV among New World bats provided a unique opportunity to explore the whether viral adaptation to new host species was repeatable and predictable and to assess the relationship between evolutionary pathways to host establishment and the timing of emergence. Positive selection occurred episodically within the evolutionary history of RV in both exposed and internal viral proteins and the adaptive changes associated with host shifts were largely unique to each virus. Moreover, our analysis demonstrated that the extent of evolution required for establishment in a new host was related to the speed of emergence.
Previous studies found mixed evidence for positive selection in some regions of G, but our study also implicated L in the establishment of new RV reservoirs (10, 12). Because our dataset encompassed more host species, viral lineages and genes, we likely had greater power to identify positive selection. Our analysis also used biologically plausible models for host switching that allowed selection to vary across genomic space and evolutionary time (15). Like all analytical methods to detect positive selection, this approach may be conservative in detecting some positively selected sites and liberal in others; however, it afforded a major advantage in the ability to detect episodic positive selection in an evolutionary history that was otherwise dominated by purifying selection (Fig. S1). Assuming that positive selection occurred during the process of host shifting, our results suggest that adaptation commonly occurred during transient periods, followed by the longer periods of purifying selection that are more typical of contemporary RV evolution (10, 12, 18). In RV, the rarity of temporally pervasive positive selection might be explained by the low efficiency of innate or adaptive host defenses against a productive viral infection in the central nervous system (18). More constant positive selection might be expected for viruses replicating in tissues with greater contact with host immune systems or reduced physiological stability.
The specific locations of amino acid sites under selection provided clues to the biological mechanisms that enabled viral establishment in new host species. First, most sites evolving under positive selection in G were found in the ectodomain. Although this region mediates viral interaction with host cells, the ability of RV to infect and replicate in all mammals studied to date suggests that entry into host cells is not a barrier for host shifts (18). Instead, viral establishment may rely on mechanisms to achieve a balance between virulence and transmission. In this regard, several regions of the ectodomain, particularly antigenic sites II and III, hold residues that can affect pathogenicity (20, 21). We found evidence for selection operating on a cluster of sites in or adjacent to antigenic site III (Table S1 and Fig. S1). Moreover, dN was elevated marginally at the site 333 of the ectodomain, including substitutions away from arginine and lysine, which can reduce or eliminate pathogenicity, disrupt cell-to-cell spread and block pathways to penetrate the central nervous system in laboratory RV strains (17, 22, 23). Although these substitutions are unlikely to have such dramatic effects on the pathogenicity of naturally circulating RVs, processes critical for the within-host progression of RV could be directly affected by these changes or indirectly affected by compensatory mutations that restore pathogenicity.
Selection was also common on internal viral regions that have minimal interaction with the host environment. These included the catalytic domain of L, the region of L that interacts with the phosphoprotein to form the RNA polymerase and the endodomain of G. One explanation for selection on these regions is that they could modulate viral transcription and replication to favor RV arrival in the salivary glands before overt host morbidity and death, enabling transmission to new hosts. Considering the diversity of bat RV reservoirs in colonial aggregation, dispersal behavior and seasonality, it seems probable that RVs vary their infection strategies across bat species (24). For example, viruses may differ in replication rate, incubation period, or ability to replicate at low temperatures in epithelial tissues, traits that might increase the probability of transmission to other species (including humans) given exposure (25).
Most of the 33 codons that were putatively linked with host adaptation experienced selection only in a handful of host shifts, and pairs of sites showed little correlated evolution (Fig. 1 and Table S2). Unique genomic routes across host shifts could result from a diverse array of functionally equivalent changes or if adaptation to each bat species required unique sets of changes. In either case, our results imply low predictability of specific sites involved in adaptation during future host shifts. This observation diverges from expectations from experimental passaging of foot-and-mouth disease virus and vesicular stomatitis virus, where the small, multifunctional genomes typical of RNA viruses appeared to promote convergent evolutionary routes to adaptation, although the environmental treatments in these studies were relatively homogenous compared with the multiple host species studied here (26, 27). On the other hand, recent studies of avian H5N1 influenza viruses demonstrated that alternative sets of amino acid changes allowed airborne transmission among ferrets, suggesting disparate genetic routes to a similar infection phenotype (28, 29). Interestingly, our results and studies in other RNA viruses suggested that replication rate could contribute to host range by altering pathogenesis and ultimately transmission (14, 30). One possibility is that replication might be manipulated through numerous processes occurring across the viral genome, opening up the potential for functionally similar but genetically distinct routes to establishment that might differ in complexity and probability. In contrast, more specific barriers such as use of novel cell receptors or evasion of immunity might constrain evolutionary pathways, particularly in surface proteins. Thus, it is possible that constraints on viral evolution might vary predictably both among viral proteins and among virus species according to the type of barriers to host-range expansion.
For RV lineages in bats, zero to five positively selected amino acid changes in G and L occurred along emergent branches, leading to new host species. Shortened evolutionary pathways to establishment might reflect ancestral changes present in donor host species that remained adaptive in the recipient host species, effectively providing an evolutionary shortcut to host adaptation (i.e., preadaptation). On the other hand, greater distance between the optimal viral genotypes for a given pair of host species could increase the number of substitutions needed for a host shift. A major question for anticipating the evolutionary and epidemiological dynamics of viral host shifts is, therefore, the degree to which variability in the evolutionary routes to adaptation affects the speed (or likelihood) of viral emergence (7). Consistent with theoretical expectations, across the series of host shifts that we studied, greater numbers of positively selected amino acid changes were associated with delayed viral establishment in new host species (Fig. 3). This suggests that viruses that undergo fewer necessary adaptive changes before establishment, either because of similarity in the adaptive landscapes of recipient and donor host species or through functionally equivalent but genetically distinct routes to adaptation, could emerge faster and with higher probability.
Despite using the best available methods and explicitly incorporating uncertainty into our statistical models, our analyses of epizootic lag time faced several challenges, including how best to define the true date of host shifts on our phylogenetic trees. We addressed this by using statistical cutoffs in our assignment of ancestral-host states to branches, assigning the dates of cross-species transmission based on different scenarios, and by including all known bat RV lineages. Although our estimates of the dates of host shifts are consistent with the first historical reports of bat rabies in the Americas during European colonization, the actual dates should be treated with caution. In particular, because we could not include undiscovered or extinct RV lineages, it is possible that some host shifts occurred more recently than we estimated (although there is no reason to suspect a systematic effect that would influence our comparative analysis). Next, because the epizootic lag times were calculated from sequence data, positively selected changes could simply have accumulated over time, with shifts to epizootic growth caused by some other factor. However, support for this scenario would also require a strong correlation between positively selected changes and synonymous substitutions, which, in turn, should have been the best predictor of epizootic lag time: neither of these criteria were supported by our analyses. Thus, although the present data cannot definitively resolve causality, our findings are most consistent with the idea that the number of positively selected sites was the underlying driver of variation in epizootic lag times. Testing this relationship in systems where experimental manipulation is possible or where ecological data exist to parameterize the epizootic lag time is an important next step. Finally, we note that although our assumption that the positive selection observed was related to cross-species transmission was supported by the timing of substitutions along branches, only controlled experiments can reveal the biological effects of these changes and whether they enable onward transmission in new host species.
In conclusion, our results showed evidence for episodic positive selection on several RV genes during the early phases of host shifts. Evolutionary routes to viral establishment shared few steps in common, suggesting that diverse genetic changes can accompany adaptation to new hosts and limiting the utility of past host shifts for predicting the evolutionary dynamics of future emergence. Importantly, variation in evolutionary routes to viral establishment might have demographic consequences: the number of positively selected changes was the best predictor of the duration of adaptive periods and, therefore, the speed of viral emergence. Identifying determinants of the extent of viral evolution needed for host shifts is therefore an outstanding question to anticipate the speed of viral establishment. These determinants could include evolutionary similarity among donor and recipient species or ecological factors, such as differences in population density, social structure, migration, or overwintering behavior (11, 24, 31, 32). Future studies linking specific viral mutations to adaptation to host biology will be important for predicting pathogen emergence, and we highlight the utility of a comparative approach that analyzes multiple, natural host shift events to guide these efforts.
We assembled datasets for the N, G, and L genes of bat RVs by supplementing published sequences with sequences generated from the virus archive of the Centers for Disease Control and Prevention (CDC) Rabies Program. The L (6,387 bp) and G (1,575 bp) sequences from this study were generated for 21 and 19 representative RV lineages (one to six isolates/lineage), respectively, from 15 bat species (SI Text and Table S5). All sequences from this study have been deposited in the GenBank database (accession nos. JQ595307–JQ595379; Table S6). Additional RV N (n = 625), G (n = 39), and L (n = 3) sequences associated with bats were collected from GenBank. For each gene, datasets were assembled that contained a maximum of 10 randomly selected but unique sequences per lineage. Final datasets comprised 30 viral lineages for N (184 unique sequences), 26 lineages for G (68 unique sequences), and 23 lineages for L (48 unique sequences). We estimated a phylogenetic tree for each dataset using five replicate ML searches in Garli Version 096b8 under substitution models selected by jModeltest, using a raccoon RV sequence as an outgroup (33, 34). The tree with the highest log likelihood was used in analyses of selection after removing the outgroup. The selection pressures at specific codon sites were estimated using the FEL method, which independently fits dN and dS to each codon position and compares the fit of these models to a null assuming dN = dS via an LRT with 1 df (16). We also used a mixed-effects model of episodic selection (MEME), which considered the dN/dS at each site as a fixed effect, while allowing for two categories of branches, those with dN/dS ≤ 1 and those with dN/dS > 1, which was treated as a random effect (15). This model was tested against a null that constrained all branches to have dN/dS < 1. Significant positive or negative selection was indicated by P values of <0.05 in the MEME analysis and P values of <0.1 in the FEL analysis because the more conservative nature of the latter test reduces the probability of type 1 errors (15, 16). As described in ref. 15, because FEL is nested within MEME, with the crucial difference being the presence of branch-to-branch variation in substitution rates in MEME, we compared the fit of the pervasive and episodic selection models at each site using LRTs with 2 df. Sites were included in later analyses if either method detected significant positive selection.
To identify bat species in which positive selection occurred, we used Bayesian phylogenetic ancestral host–state reconstruction using Bayesian Evolutionary Analysis by Sampling Trees (BEAST) Version 1.7 (35, 36). We extended a previously published analysis by allowing host shifts to occur asymmetrically between host species, by including viral lineages from Central and South American bat species and, most importantly, by integrating information from the N, G, and L genes in a joint phylogenetic analysis (11). For each of the 30 host-associated lineages, we selected a maximum of three representative isolates, preferentially choosing those isolates for which we had sequences from multiple genes. Thus, our dataset comprised 87 isolates with a total of 86, 58, and 44 sequences for N, G, and L, respectively. Each alignment was treated as a separate data partition, allowing us to estimate a consensus evolutionary history of bat rabies and the most probable host states along branches (see SI Text for analytical details).
We assembled 13 datasets of N gene sequences for which a minimum of 20 sequences (mean, 45; range, 20–82) were sampled over at least 10 y (mean, 22.3; range, 10–30 y). N was chosen because it was the most thoroughly sampled gene in terms of the number and temporal range of sequences. Bayesian skyline plots (BSPs) were estimated in BEAST using substitution and molecular clock models that were customized for each lineage (Table S3). We compared models assuming constant population sizes, exponential growth, and logistic growth using the stepping-stone method to calculate the marginal likelihood of each demographic model, enabling the use of Bayes factors (BFs) for hypothesis testing (37). Rejection of the constant population size model (BF > 3) by the exponential or logistic growth model was considered evidence of population growth. A single exception was made for the virus maintained by Desmodus rotundus (DrV), which had a complex demographic history that was poorly captured by the parametric models relative to the BSP (Fig. 2 and Table S3). When viral population growth was supported, we estimated the beginning of the growth phase using parametric two-epoch models, characterized by two historical periods of constant but distinct viral population sizes and a parameter describing the transition time between them (38, 39). Priors for demographic parameters were informed from the results of the BSPs. The uniform prior on the molecular clock was constrained to the 95% bounds of that parameter from the corresponding BSP analysis; the 95% bounds of population sizes at the beginning and end of the BSP were used as the bounds of the 1/x prior distributions for each constant size period; the lower limit of the transition time prior was the lower 95% bound of the time since the most recent common ancestor of the lineage and the upper limit was 5–15 y before the most recently sampled virus. Thus, we effectively fit the two-epoch model to the observed BSP for each lineage but verified that posterior distributions were not strongly impinged by our prior choices and used more diffuse priors when necessary (38). Demographic inference combined three to four replicate runs of 100 million generations after discarding the first 20% of each as burn-in.
We thank Ivan Kuzmin for providing some of the oligonucleotide sequences, Sergei Kosakovsky Pond for suggestions on detection of positive selection, and Eric Crandall for help with the two-epoch demographic models. We thank many colleagues in the US state health departments and abroad for submission of samples for diagnostic confirmation and viral characterization. D.G.S. was supported by a Dissertation Completion Award from the University of Georgia and National Science Foundation Grant DEB-1020966.
Author contributions: D.G.S. designed research; D.G.S. performed research; D.G.S. analyzed data; and D.G.S., S.M.A., A.V.-V., and C.E.R. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. E.C.H. is a guest editor invited by the Editorial Board.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. JQ595307–JQ595379).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1203456109/-/DCSupplemental.