A CRISPR set of materials
CRISPR technology is best known as a gene editing tool. English et al. developed a group of stimuli-responsive hydrogels to respond to the programmable nuclease Cas12a (see the Perspective by Han et al.). The materials undergo molecular to macroscopic changes after Cas12a-dependent cleavage of double- or single-stranded DNA integrated into the gel. The authors show controlled release of particles linked to or imprisoned within the DNA, degradation of a gel with DNA solely forming the cross-links, and permeabilization of a gel with DNA partially forming the cross-links. These tools allow for the production of materials that release encapsulated nanoparticles and cells, act as degradable fuses, and enable remote radio-frequency identification signaling.
Abstract
Stimuli-responsive materials activated by biological signals play an increasingly important role in biotechnology applications. We exploit the programmability of CRISPR-associated nucleases to actuate hydrogels containing DNA as a structural element or as an anchor for pendant groups. After activation by guide RNA–defined inputs, Cas12a cleaves DNA in the gels, thereby converting biological information into changes in material properties. We report four applications: (i) branched poly(ethylene glycol) hydrogels releasing DNA-anchored compounds, (ii) degradable polyacrylamide-DNA hydrogels encapsulating nanoparticles and live cells, (iii) conductive carbon-black–DNA hydrogels acting as degradable electrical fuses, and (iv) a polyacrylamide-DNA hydrogel operating as a fluidic valve with an electrical readout for remote signaling. These materials allow for a range of in vitro applications in tissue engineering, bioelectronics, and diagnostics.
Microbial clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) adaptive immune systems contain RNA-guided endonucleases capable of multiple-turnover nucleic acid hydrolysis (1, 2). Because of their specificity and programmability, CRISPR-Cas enzymes have been exploited as efficient genome-editing tools (3) and in nucleic acid diagnostic applications such as SHERLOCK (4, 5) and DETECTR (2).
Biologically responsive materials are important for biotechnology applications, including the fabrication of scaffolds for tissue engineering (6), the actuation of microfluidic valves (7), and the detection of analytes in sensors (8, 9). DNA-responsive hydrogels are well suited to interface with synthetic DNA constructs or naturally occurring extracellular DNA (10). Current DNA-responsive hydrogels typically rely on strand displacement or structural changes in DNA cross-linkers (11, 12), which require high concentrations of DNA triggers for actuation. Adapting such DNA hydrogels for activation with new trigger sequences involves the modification of nucleic acid components, which can conflict with the structural requirements (for example, length or secondary structure) imposed by the material. This limits the programmability of these systems and highlights the need for a strategy that uncouples structural and sensing constraints in DNA-based materials.
We use Cas enzymes to control the properties of hydrogels with integrated DNA components at multiple scales and in a modular fashion and thereby eliminate the need to encode target-sequence specificity into the gel structure. Our material platform is able to induce changes in hydrogels in response to user-defined target nucleic acid sequences by replacing a single component, a CRISPR guide RNA (gRNA) that governs sequence-specific Cas activation. We engineered different DNA-based materials to demonstrate a variety of nuclease-mediated responses, including the release of small molecules, enzymes, nanoparticles (NPs), and live cells, as well as the modulation of bulk electrical and permeability properties of DNA hydrogels for sensing and diagnostics (fig. S1).
Cas12a has both targeted and collateral deoxyribonuclease activities
The Cas12a from Lachnospiraceae bacterium ND2006 (1, 2) displays a specific cleavage activity toward double-stranded DNA (dsDNA) fragments matching the gRNA spacer sequence (targeted cleavage) and subsequent indiscriminate single-stranded DNA (ssDNA) hydrolysis activity (collateral cleavage, fig. S2). Once the Cas12a-gRNA complex binds and cleaves its dsDNA target, the collateral cleavage of nearby ssDNA by Cas12a is highly efficient (~1250 turnovers per second) (2). To demonstrate the target programmability of CRISPR gels, we tested gRNAs to detect fragments of the mecA antibiotic-resistance gene of methicillin-resistant Staphylococcus aureus (MRSA). We selected MRSA as a clinically relevant model to demonstrate the target programmability of CRISPR gels, given the high concentration of extracellular DNA in MRSA biofilms (13).
We first validated the collateral cleavage activity of Cas12a-gRNA in solution (14). Cleavage of an ssDNA reporter containing a quenched fluorophore by Cas12a in response to mecA dsDNA triggers confirmed the performance of the MRSA1 gRNA (fig. S3). Cas12a programmed with this gRNA detected its cognate trigger down to ~16 pM in solution (fig. S4) and was selected for subsequent experiments. Sequence mismatches between the gRNA and trigger reduced the collateral cleavage rate, consistent with previously reported patterns (2, 15). Nonspecific activation of Cas12a by a scrambled dsDNA sequence was over 100-fold lower than that by the mecA trigger (fig. S4). Cas12a’s high dsDNA target specificity and rapid nonspecific ssDNA collateral cleavage activity make it an ideal candidate for modulating a wide range of physical and mechanical hydrogel properties (fig. S1).
Programmable materials capable of the controlled release of soluble compounds, as well as encapsulated cells, have broad utility in various therapeutic and research applications (16–19). Because of the diversity of DNA-based hydrogels in the literature (table S1), we selected material formulations that span a wide range of physical properties to demonstrate that they could be actuated by Cas proteins (table S2): (i) poly(ethylene glycol) (PEG) hydrogels with covalently bound nonstructural DNA pendants that are released by Cas12a nuclease activity without degrading the overall hydrogel structure; (ii) acrylamide hydrogels with structural ssDNA cross-links that could be cleaved by Cas12a, resulting in bulk gel degradation as well as particle or cell release; and (iii) conductive hydrogels loaded with carbon black (CB) that were cleaved from the surface of electrodes by Cas12a, thus behaving like an electronic fuse.
Collateral Cas12a activity releases ssDNA-anchored cargos from hydrogel matrices
The targeted dsDNA cleavage activity of Cas12a can be used to preferentially release anchored cargos with near-single turnover (fig. S5); however, we focused on the collateral ssDNase activity of the enzyme, as it allows for the efficient transduction of external stimuli into changes in material properties through catalytic signal amplification. To illustrate the programmable actuation of materials using Cas12a, we covalently tethered a fluorophore (Cy3) into PEG hydrogels through an ssDNA linker (Fig. 1A) and monitored its release into solution (fig. S6) upon Cas12a-induced cleavage. The Cas12a-gRNA complex was insufficient to catalyze cargo release; however, introduction of the mecA dsDNA trigger initiated the hydrolysis of ssDNA anchors (Fig. 1B and fig. S7). By contrast, a randomly permutated version of the mecA dsDNA (scrambled control) failed to do so. We then used horseradish peroxidase (HRP) enzyme as a model for larger biomolecule payloads, demonstrating that biological function was preserved after immobilization in hydrogels using ssDNA anchors and subsequent release by Cas12a (Fig. 1C). Within 10 min of exposure to a low-concentration (10 nM) dsDNA stimulus, we detected sufficient HRP activity in the supernatant for a visual readout (Fig. 1C, inset). Further incubation allowed us to routinely discriminate trigger and scrambled dsDNA down to 100 pM (fig. S8). These experiments are consistent with the efficient ssDNase activity of activated Cas12a (2).
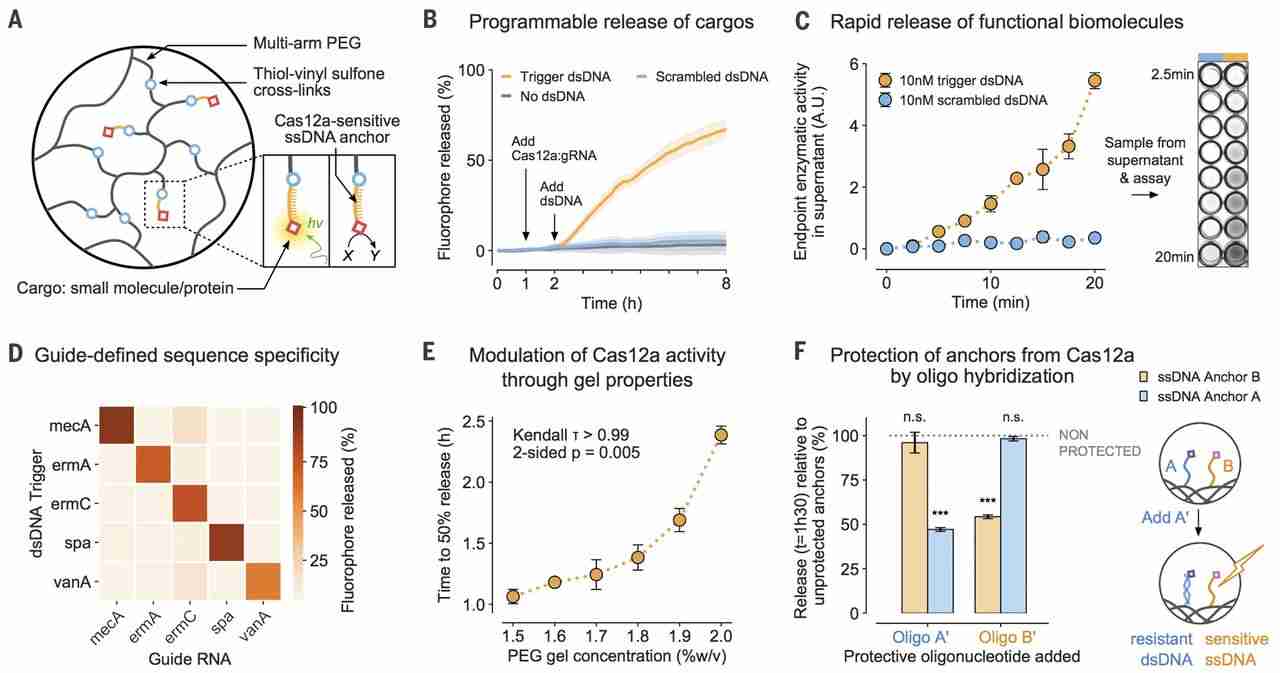
(A) ssDNA acts as a cleavable linker for attaching payloads to an inert PEG matrix. hν, light energy. (B) Release of a tethered fluorophore by Cas12a is initiated only upon introduction of a specific dsDNA trigger and not a scrambled dsDNA control sequence. (C) Functional enzymes can be anchored into the hydrogel and released by Cas12a in sufficient quantities for visual detection in an HRP activity assay within minutes. A.U., arbitrary units. (D) Activation of Cas12a and fluorophore release (t = 8 hours) is defined by the complementarity between a dsDNA sequence and the gRNA of Cas12a. (E) Cross-linking density of the PEG hydrogels modulates the release rate of the cargo by Cas12a. The correlation was analyzed using a Kendall rank test. (F) Prehybridization of the ssDNA linkers with a matching oligonucleotide selectively reduces the release rate of molecules anchored in the gel (observed at t = 1.5 hours). The means were compared with independent samples that were not preprotected with oligonucleotides (gray). Differences in the means of the test conditions and the unprotected controls were analyzed using a t test [Bonferroni-adjusted α = 0.0125, P values: not significant (n.s.) P > 0.05, ***P < 0.0001]. All plots show mean ± SD for n ≥ 3 replicates.
To demonstrate that changes to the gRNA were sufficient to entirely reprogram the target responsiveness of the material, we designed gRNAs to target a panel of genes involved in S. aureus antibiotic-resistance mechanisms (fig. S9). These include the antibiotic-resistance genes ermA and ermC (20, 21), the virulence factor gene spa (22), and the vancomycin-resistance gene vanA (23). Out of 25 combinations of gRNAs and dsDNA, those in which the sequence of the trigger matched the gRNA resulted in substantially higher fluorophore payload release from the hydrogel matrix (Fig. 1D). These results correlated to similar observations of the reactions performed in solution (fig. S10) and suggest that different gRNA-trigger pairs activate Cas12a to different extents (2).
For many applications, the rate at which a molecule is delivered from a carrier conveys important biological information (16). The speed of CRISPR-mediated hydrogel actuation corresponds to the amount of input dsDNA (fig. S11); conversely, for a given level of input, the response dynamics can be hard-coded into the system by altering the properties of the starting material. For example, pore size is expected to alter the mobility of macromolecules in polymer networks (24). On the basis of our macroscopic observations of programmed anchor hydrolysis (fig. S12), we hypothesized that this could be used to further tune the relationship between dsDNA input and Cas12a-mediated response. By modulating the cross-linking density of a PEG-DNA hydrogel and measuring the rate of fluorophore release by Cas12a-gRNA, we established another strategy by which the behavior of the CRISPR-responsive material could be controlled (Fig. 1E and figs. S13 and S14).
In addition to controlling global dynamics of ssDNA cleavage through bulk material properties, we capitalized on the sequence-defined, addressable nature of the ssDNA linkers and the selectivity of the collateral cleavage activity of Cas12a for ssDNA over dsDNA (1, 2). We attached two different fluorophores (Cy3 and 6-FAM) into PEG hydrogels with distinct ssDNA linkers and preprogrammed the differential sensitivity of one linker over the other to Cas12a collateral degradation by hybridizing it with a complementary blocking strand in situ. Whereas the release of the unprotected fluorophore was unaffected, the speed of release of the hybridized reporter was markedly reduced (Fig. 1F and fig. S15).
Collateral Cas12a activity alters the large-scale mechanical properties of DNA hydrogels
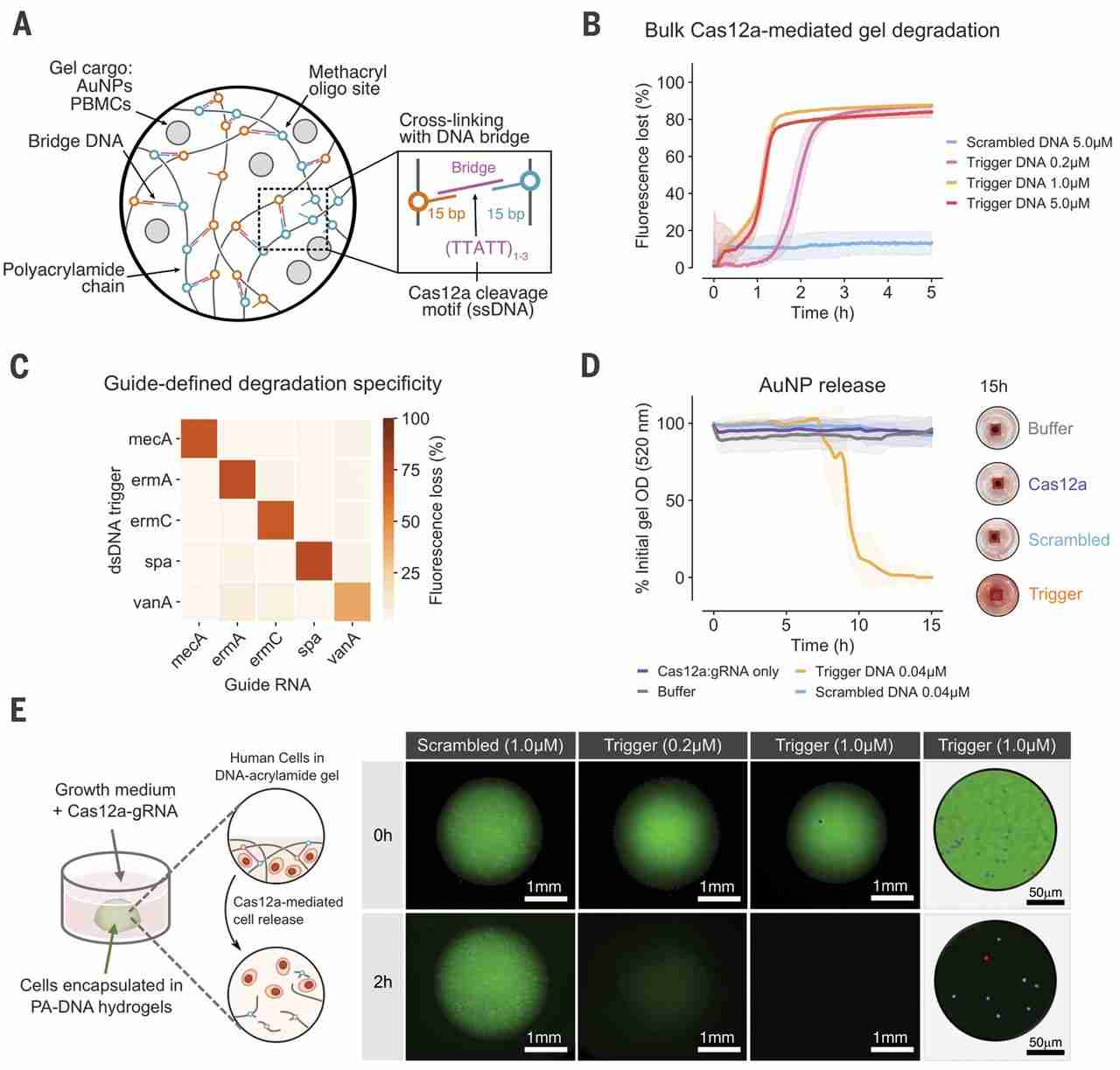
The high catalytic efficiency of dsDNA-activated Cas12a-gRNA (kcat/Km ~ 1.7 × 109 s−1 M−1) (2) makes it well suited for converting dsDNA signals into bulk material changes. To demonstrate this, we designed DNA cross-linked polyacrylamide (PA) hydrogels (9, 25) by separately incorporating two noncomplementary oligonucleotides into PA chains (fig. S16). We then cross-linked the PA-DNA precursors using an oligonucleotide strand that forms bridges between the PA-DNA chains. These cross-links contained single-stranded, AT-rich Cas12a collateral cleavage sites (Fig. 2A). In these hydrogels, degradation of DNA cross-links physically disrupts the polymer networks (fig. S1B) (24, 26).
(A) ssDNA bridges lock DNA-functionalized PA chains into a 3D network. (B) Cas12a-mediated degradation of PA-DNA gels stained with EvaGreen intercalating DNA dye. (C) Degradation of gel with 25 combinations of gRNAs and dsDNA triggers and comparison of signals after 12 hours. (D) Release of AuNPs from 7% (w/v) PA-DNA gels using Cas12a collateral cleavage, tracked by measuring gel optical density. The Cas12a-gRNA and dsDNA trigger were encapsulated in the gel with the AuNPs (concentrations shown include supernatant volume). (E) Sequence-specific degradation of PA-DNA gels leads to the release of encapsulated nonadherent PBMCs. Cells were stained before encapsulation using calcein acetoxymethyl ester (blue) and ethidium homodimer (red), and gels were labeled with a fluorescein-functionalized ssDNA bridge. Postdegradation live-dead staining is shown in figs. S24 and S25. All plots show mean ± SD for n ≥ 3 replicates.
The Cas12a-induced degradation of PA-based CRISPR gels was initially evaluated with a DNA-intercalating dye to label bridge sequences in PA-DNA gels and track gel integrity. The bridges were degraded upon exposure to gRNA-Cas12a and trigger dsDNA, as revealed by the dissipation of gel fluorescence at rates dependent on trigger concentration (Fig. 2B). Compared with experiments performed in solution (fig. S4), gel degradation appeared more robust to the introduction of sequence mismatches between the gRNA and dsDNA trigger (fig. S17). Using fluorescein isothiocyanate–dextran particles physically entrapped in the hydrogel, we also visualized the degradation of millimeter-scale PA-DNA hydrogels (fig. S18).
Programmable degradation of PA-DNA hydrogels was assessed by testing 25 combinations of different gRNAs and dsDNA triggers. Consistent with the nondestructive cargo-release experiments (Fig. 1E), PA-DNA hydrogel degradation occurred only when the gRNA and dsDNA sequences were complementary (Fig. 2C and fig. S19), demonstrating Cas12a-gRNA’s ability to discriminate between inputs.
Though biomolecules can be tethered to materials through well-defined, single linkers, physical entrapment in a polymer matrix represents a more general strategy to control the release of larger payloads. We tested the release of NPs by encapsulating PEG-coated gold NPs (AuNPs) (figs. S16C and S20) in PA-DNA hydrogels. Loading gels with both Cas12a-gRNA and a dsDNA trigger led to total NP release through Cas12a activation and gel degradation, whereas gels loaded with a scrambled dsDNA trigger showed no significant release of AuNPs relative to a buffer-only background (Fig. 2D and figs. S21 and S22). This was consistent with the disruption of the percolated network upon cross-link cleavage (27, 28).
The complex interactions between cells and surrounding materials have implications for tissue engineering and other therapeutic applications. We reasoned that Cas12a-gRNA would have the capacity to modify the extracellular matrix of cells encapsulated in DNA materials in response to predefined cues. We tested the reversible encapsulation of human primary peripheral blood mononuclear cells (PBMCs) in PA-DNA hydrogels by exposing the gels to activated gRNA-Cas12a. Complete gel degradation and cell release were observed within 2 hours in the presence of 1 μM dsDNA trigger (Fig. 2E and fig. S23), without compromising cell viability (figs. S24 and S25). Conversely, gels exposed to a scrambled dsDNA control remained intact in the same time frame. These results demonstrate that NPs and live-cell payloads can be immobilized in biocompatible hydrogels and released upon addition of trigger dsDNA sequences without the need for hydrogel redesign to accommodate different input signals.
Conductive DNA-based materials act as Cas12a-actuated electronic fuses
We used Cas12a to modulate the attachment of a conductive DNA-based hydrogel to an electrode surface to act as an electrical fuse triggered by specific DNA sequences (Fig. 3A and fig. S1C). A conductive, biologically responsive hydrogel may be desirable for a variety of sensing and diagnostic applications when the direct interface to electrical devices (such as analog circuits and microcontrollers) is required (29). These conductive, self-assembled materials consisted of ssDNA networks cross-linked with CB-conductive NPs (CB-DNA gels). CB is composed of spherical particles (30) containing graphitic-like domains (31) and is widely used in industrial applications to impart electrical conductivity to polymers (32); in this study, we used it as a conductive cross-linker in hydrogels. CB-DNA gels were synthesized through thermal melting of dsDNA followed by cooling in the presence of CB NPs. This leads to the strong, noncovalent association of the aromatic DNA nucleotides with nearby CB graphitic surfaces (33) through hydrogen bonding and π-π stacking interactions (34–36). In these hydrogels, DNA behaves as the main structural component, linking CB particles together to form a three-dimensional (3D) network (34, 37).
(A) Schematic of the experimental workflow. (B) Degradation kinetics of CB-DNA gels with increasing concentrations of DNA trigger (n = 5 electrodes per condition). We inspected replicates at hourly intervals to record detachment of gels from interdigitated electrodes (IDEs). PET/Ag, Polyethylene terephthalate/silver. (C) Electrical resistance across the IDEs in (B) after removal from the reaction upon detachment of the CB-DNA gel or after 24 hours if no detachment occurred. Measurements before and after reaction were compared using a Kruskal-Wallis (K-W) test (before: P = 0.7, after: P = 0.0003) and Dunn’s post hoc test (before: all > 0.99; after: P < 0.05 for both 1.0 μM and 0.5 μM versus controls, P > 0.05 otherwise). (D) A representative image of a gel in the reaction mix detached from its IDE (top) and microscopy images of the IDEs in (B) after removal from the reaction (bottom). Images are quantified in fig. S23A. Scram., scrambled.
We hypothesized that cleavage of the ssDNA at the electrode-material interface by Cas12a would disrupt the conductive path. To test this system, CB-DNA droplets were spotted onto printed interdigitated silver electrodes and lyophilized (Fig. 3A). Before Cas12a-mediated degradation, lyophilized CB-DNA hydrogels showed conductivities comparable to that reported for graphene-DNA gels of similar compositions (~4 mS/cm) (34). After initial electrical testing, CB-DNA gels were incubated in a solution containing Cas12a-gRNA and dsDNA triggers. We visually monitored the CB-DNA hydrogels during Cas12a-mediated detachment (Fig. 3B), tested for conductivity (Fig. 3C), and imaged the electrodes (Fig. 3D and fig. S26A) with increasing concentrations of dsDNA inputs. Cas12a-gRNA with 500 nM dsDNA trigger was able to completely detach 60% of the hydrogels from electrodes in 10 hours and 100% of hydrogels after 20 hours. Incubation with a higher dsDNA trigger concentration (1 μM) led to CB-DNA detachment from 100% of electrodes within 10 hours. Complete detachment resulted in an opening of the circuit across the electrode, whereas partial detachment of the CD-DNA hydrogels at lower dsDNA trigger concentrations led to intermediate conductivities (Fig. 3, C and D, and fig. S26). Exposure of electrodes with CB-DNA gels to an ssDNA-specific nuclease resulted in a similar response, confirming that detachment was a consequence of Cas12a activation and ssDNA hydrolysis (fig. S26, D and E). This inexpensive CB-DNA gel formulation provides a direct link between dsDNA triggers and electrical outputs.
Cas12a-controlled hydrogel formation in a paper fluidic device enables diagnostic readouts
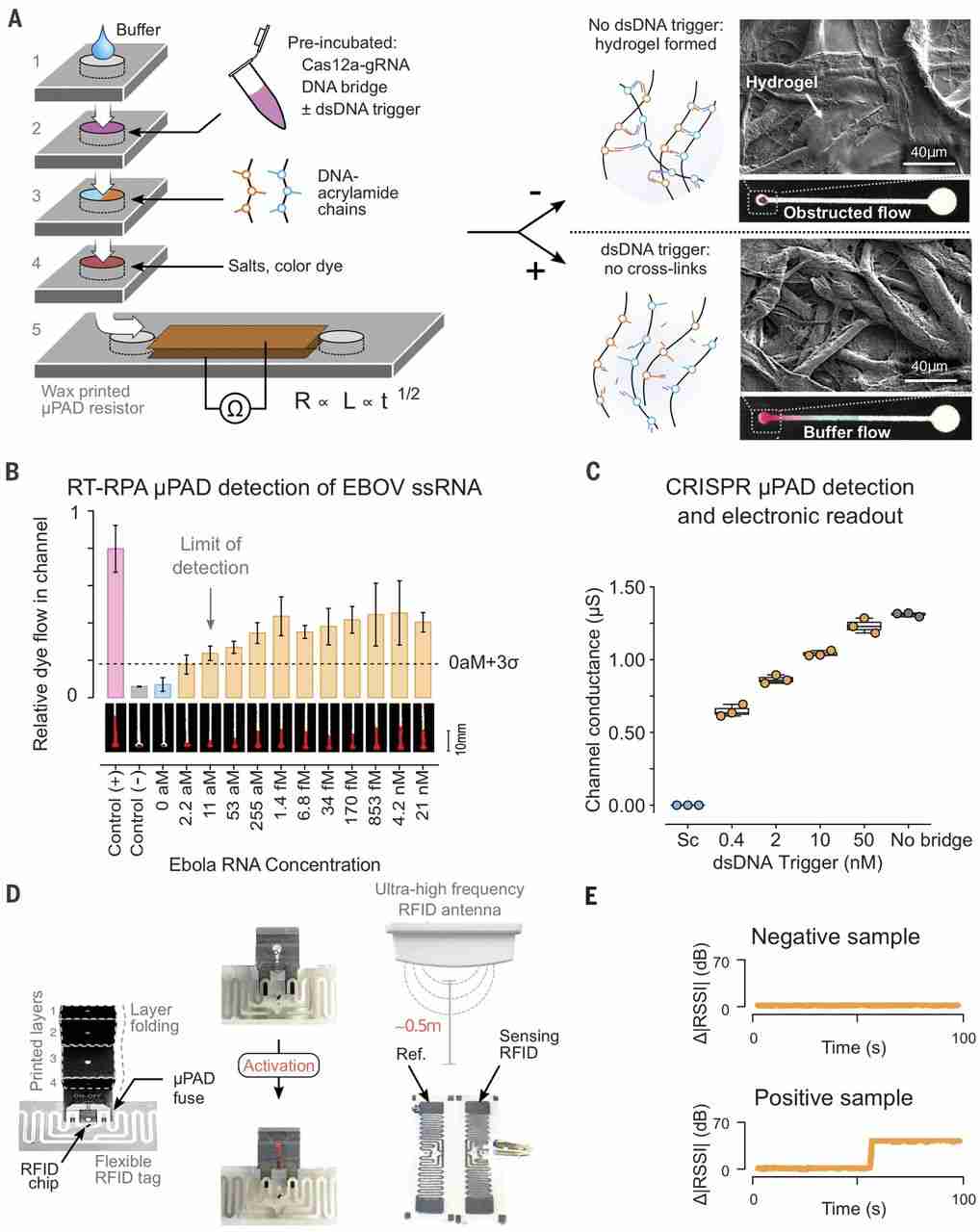
We used a tunable PA-DNA hydrogel to control the permeability and electrical readout of a paper-based microfluidic device (Fig. 4). Paper-based technologies have shown promise for point-of-care diagnostics, as they are low cost, equipment-free, and easy to use (38, 39). Our device (Fig. 4A and fig. S24) expands on the concept of microfluidic paper-based analytical devices (μPADs) that rely on the capacity of hydrogels to obstruct flow through porous channels (40).
(A) Schematic of the stackable μPAD design (40) modified for operation with CRISPR gels and electrical readout. Layers 1 to 4 contain hydrophilic regions that form a continuous channel on folding and feed into a lateral flow channel in layer 5. The channel in layer 5 was covered with conductive tape to measure conductivity as a function of buffer wicking. In the presence of target trigger, Cas12a cleavages the DNA linker, preventing hydrogel cross-linking in the channel and enabling flow. The inset shows SEM images of channels with (top) and without (bottom) cross-linked hydrogel. (B) End point measurements (t = 5 min) of the colorimetric-coupled RT-RPA μPAD hydrogel detection system for different concentrations of ssRNA Ebola virus (EBOV) input (mean ± SD, n = 3 μPADs). Representative images of μPAD channel flow are shown. The positive control corresponds to flow with no ssDNA bridging strand in the preincubation reaction, and the negative control corresponds to flow with an undigested ssDNA bridging strand. Student’s t test P = 0.0057 for differences in the means of the 0- and 11-aM ssRNA samples. (C) End point measurements (5 min) of resistance across the channel for different concentrations of dsDNA MRSA trigger input after a 4-hour predigestion step. Sc = 50 nM scrambled dsDNA. (D) Schematic illustrating the integration of the paper-fluidic device with an RFID flexible tag. Cas12a activation in the preincubation step results in the short-circuiting of an interdigitated electrode arrangement in the loop RFID tag, thereby altering the received signal strength indicator (RSSI) compared with a reference tag (Ref.). An increase in absolute RSSI difference between the tags is indicative of Cas12a activation by a dsDNA trigger. (E) Representative signal traces for positive and negative results in the experimenter-blinded trial of the RFID μPAD device (full set in fig. S29). Samples containing either 0 aM (negative) or 11 aM (positive) EBOV ssRNA trigger were amplified by RT-RPA, incubated with the ssDNA gel bridging strand and Cas12a-gRNA for 4 hours, and assayed on a μPAD-RFID device.
The layers of the device were folded to create a multilayered structure in which the hydrophilic regions are topologically aligned. Capillary-driven flow through the device terminated in a fifth layer where the output was measured (Fig. 4A and fig. S27). In this system, an intermediary layer contains PA-DNA gel precursors (Ps-X and Ps-Y) that, when mixed with ssDNA cross-linker, form a hydrogel in the paper channels (41, 42). The extent of gel formation, and therefore the rate of buffer flow, is dependent on the extent of degradation of the ssDNA gel cross-linker during a preincubation step. The activation of Cas12a can be confirmed by adding a fluorescent ssDNA reporter during this step (fig. S28). By degrading the cross-linker by using Cas12a, we were able to couple the level of buffer flow to the concentration of dsDNA trigger added to a Cas12a reaction incubated for 4 hours.
When nonspecific dsDNA trigger is present during preincubation, ssDNA cross-linkers are not cleaved, allowing for hydrogel assembly in the microchannel (Fig. 4A). Conversely, in the presence of a specific dsDNA trigger, unimpeded flow can be visually detected by adding dyes to the μPAD channel. We found the rate of buffer flow through a μPAD to be inversely related to the concentration of an MRSA dsDNA trigger. Using this visual output, we were able to detect dsDNA concentrations down to 400 pM (fig. S29).
To optimize our CRISPR-μPAD for field diagnostic applications, we used reverse transcription (RT) to expand the range of detectable biomarkers to RNA and coupled the RT to an isothermal amplification [recombinase polymerase amplification (RPA)] step to improve the limit of detection. We used RT-RPA followed by a μPAD readout to detect synthetic Ebola genomic RNA (43) down to 11 aM (Fig. 4B), a sensitivity matching other state-of-the-art CRISPR-based diagnostics (fig. S30) (2, 4, 5). This approach is promising for point-of-care diagnostics and has overall better performances in terms of sensitivity, portability, and cost than other molecular diagnostics (table S3).
Visual readouts of buffer flow are commonly used, yet they are difficult to couple to downstream hardware for data processing. To overcome this limitation, we modified the CRISPR-actuated fluidic system to read buffer flow as an electric signal: the microfluidic channel in the final layer was sandwiched between two electrodes and connected to an ohmmeter (Fig. 4A and fig. S27B). Electrical conductivity between the electrodes relied on electrolytes provided by the flowing buffer and was directly correlated to the buffer penetration length in the μPAD channel (fig. S31A) (44). Using this approach, sub-nanomolar concentrations of dsDNA trigger were successfully detected at a 5-min end point, without DNA amplification, demonstrating the potential of the CRISPR μPAD for embedded sensor applications (Fig. 4C). Nonspecific dsDNA trigger did not activate Cas12a, thus leaving the electrical circuit open (Fig. 4C and fig. S31). We were able to reduce the preincubation time required to observe a signal to 1 hour by tuning the properties of the acrylamide precursors (fig. S31B).
The wireless, decentralized logging of individual clinical tests during infectious disease outbreaks could address challenges with record keeping and logistics. To integrate CRISPR-Cas reactions with electronic monitoring systems through hydrogel actuation, we incorporated a wireless radio-frequency identification (RFID) module into the μPAD. The original design was modified such that buffer flow would short-circuit an interdigitated silver electrode, thereby modulating the efficiency of signal transmission by a flexible RFID tag (Fig. 4D and fig. S31). We then conducted an experimenter-blinded trial consisting of 12 samples (containing either 11 or 0 aM Ebola ssRNA amplified by RT-RPA) divided across three geographic locations (Fig. 4E and fig. S32). The experimenter preincubated the samples with Cas12a and Ebola-specific gRNA for 4 hours and then recorded the RFID-μPAD signals over the course of 2 min. Buffer flow through the μPAD in Ebola-positive samples caused short-circuiting of the RFID tag antenna, which was detected in real time as a change in the signal strength compared with an unmodified reference RFID tag (Fig. 4E). All positive and negative samples were correctly assigned using the RFID-μPAD (fig. S32).
We have demonstrated several strategies to interface biological signals with materials that combine the inherent programmability of Cas enzymes with hydrogel systems. These strategies offer control over a variety of complex behaviors and properties, including the release of molecules, NPs, and live cells, as well as bulk hydrogel degradation, electronic signal transduction, and microfluidic valve actuation. By exploiting the enzymatic properties of Cas12a, we have designed a platform that improves on hydrogel programmability and versatility, as only the gRNA molecule needs to be changed to allow hydrogel response to a user-defined DNA sequence. The catalytic activity of Cas12a improves sensitivity compared with DNA-responsive hydrogels requiring stochiometric amounts of DNA triggers for material activation. Finally, we demonstrate various forms of output that expand the capabilities of CRISPR-responsive materials and enhance existing biomaterial-based approaches for tissue engineering, molecular diagnostics, and bioelectronic interfaces with programmable readouts.
Supplementary Materials
science.sciencemag.org/content/365/6455/780/suppl/DC1
Materials and Methods
Figs. S1 to S32
Tables S1 to S4
This is an article distributed under the terms of the Science Journals Default License.



{kind=link}
{kind=link}